
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
CURRENT REPORT
Pursuant to Section 13 OR 15(d) of The Securities Exchange Act of 1934
Date of Report (Date of earliest event reported):
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of incorporation) |
(Commission File Number) |
(IRS Employer Identification No.) | ||
| (Address of principal executive offices) | (Zip Code) | |||
Registrant’s telephone number, including area code: (
N/A
(Former name or former address, if changed since last report.)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Trading |
Name of each exchange on which registered | ||
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§ 230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§ 240.12b-2 of this chapter).
Emerging growth company
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
| Item 7.01 | Regulation FD Disclosure. |
On August 22, 2023, Fulcrum Therapeutics, Inc., or Fulcrum, issued a press release announcing that the U.S. Food and Drug Administration lifted the clinical hold on the Investigational New Drug application for FTX-6058 for the potential treatment of sickle-cell disease. In connection therewith, Fulcrum posted a program update presentation, or the Presentation, that provides more information about the amended protocol and planned Phase 1b trial of FTX-6058 on the Investor Relations section of its website at www.fulcrumtx.com. A copy of the Presentation entitled “FTX-6058 for Sickle Cell Disease Program Update” is furnished as Exhibit 99.1 hereto and incorporated by reference herein. Fulcrum expressly disclaims any obligation to update the Presentation, or any other information posted on or available through its website, and cautions that the information set forth therein is only accurate as of the date indicated on such materials. The inclusion of any data or statements in the Presentation (or available on or through Fulcrum’s website) does not signify that such information is considered material.
The information in this Item 7.01, including Exhibit 99.1, shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or otherwise subject to the liabilities of that section, nor shall it be deemed incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as expressly set forth by specific reference in such a filing.
| Item 9.01 | Financial Statements and Exhibits. |
| (d) | Exhibits |
The following exhibit is furnished herewith:
| 99.1 | Corporate Presentation - FTX-6058 for Sickle Cell Disease Program Update | |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL document) | |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| FULCRUM THERAPEUTICS, INC. | ||||||
| Date: August 22, 2023 | By: | /s/ Curtis Oltmans | ||||
| Name: | Curtis Oltmans | |||||
| Title: | Chief Legal Officer | |||||

Exhibit 99.1 FTX-6058 f o r S i c k l e C e l l D i s e a s e Program Update

F U L C R U M T H E R A P E U T I C S Disclaimer and Notice This presentation contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 that involve substantial risks and uncertainties. All statements, other than statements of historical facts, contained in this presentation are forward-looking statements, including express or implied statements regarding resuming clinical development of FTX-6058; the Phase 1b clinical trial design and number of subjects in each cohort as well as the number of cohorts; and the effects of the revised inclusion and exclusion criteria; and enrollment in such trial; among others. The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. Any forward-looking statements are based on management’s current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in, or implied by, such forward-looking statements. These risks and uncertainties include, but are not limited to, risks associated with conducting clinical trials; Fulcrum’s ability to continue to advance its product candidates in clinical trials; initiating and enrolling clinical trials on the timeline expected or at all; obtaining and maintaining necessary approvals from the FDA and other regulatory authorities; replicating in clinical trials positive results found in preclinical studies and/or earlier-stage clinical trials; obtaining, maintaining or protecting intellectual property rights related to its product candidates; managing expenses; managing executive and employee turnover, including integrating a new CEO and CFO; and raising the substantial additional capital needed to achieve its business objectives, among others. For a discussion of other risks and uncertainties, and other important factors, any of which could cause Fulcrum’s actual results to differ from those contained in the forward-looking statements, see the “Risk Factors” section, as well as discussions of potential risks, uncertainties, and other important factors, in Fulcrum’s most recent filings with the Securities and Exchange Commission. In addition, the forward-looking statements included in this presentation represent Fulcrum’s views as of the date hereof and should not be relied upon as representing Fulcrum’s views as of any date subsequent to the date hereof. Fulcrum anticipates that subsequent events and developments will cause Fulcrum’s views to change. However, while Fulcrum may elect to update these forward-looking statements at some point in the future, Fulcrum specifically disclaims any obligation to do so. 2
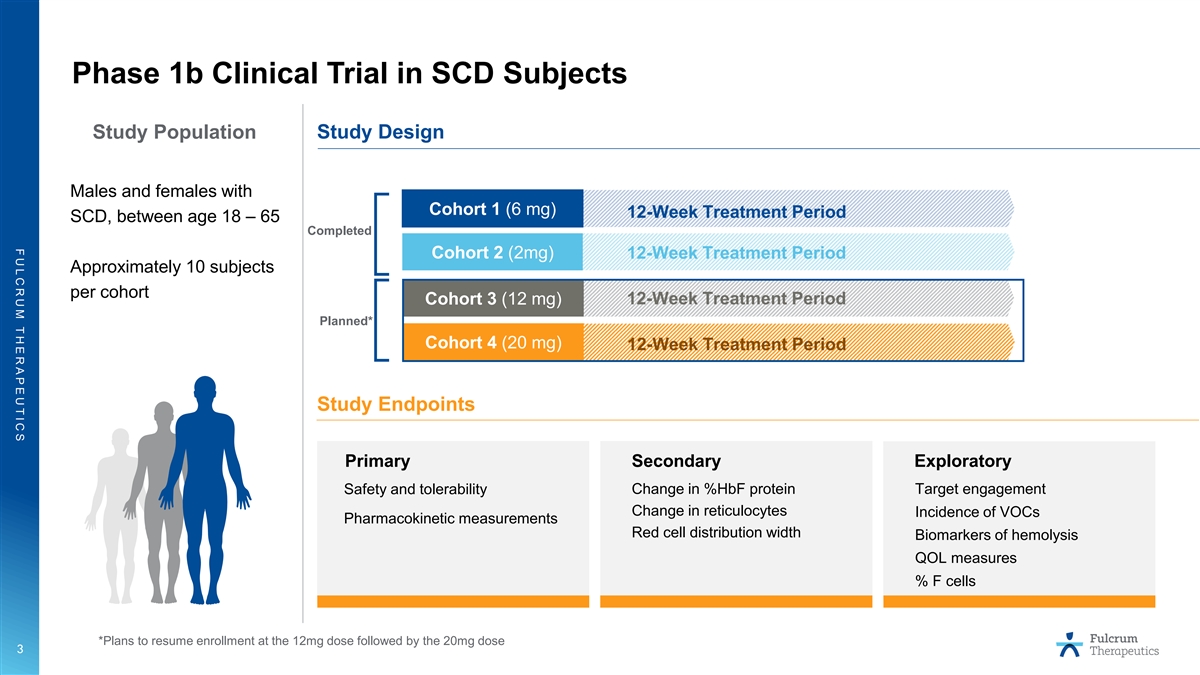
F U L C R U M T H E R A P E U T I C S Phase 1b Clinical Trial in SCD Subjects Study Population Study Design Males and females with Cohort 1 (6 mg) 12-Week Treatment Period SCD, between age 18 – 65 Completed Cohort 2 (2mg) 12-Week Treatment Period Approximately 10 subjects per cohort Cohort 3 (12 mg) 12-Week Treatment Period Planned* Cohort 4 (20 mg) 12-Week Treatment Period Study Endpoints Primary Secondary Exploratory Change in %HbF protein Target engagement Safety and tolerability Change in reticulocytes Incidence of VOCs Pharmacokinetic measurements Red cell distribution width Biomarkers of hemolysis QOL measures % F cells *Plans to resume enrollment at the 12mg dose followed by the 20mg dose 3

F U L C R U M T H E R A P E U T I C S Initial FTX-6058 Data Demonstrates Dose-dependent Increases in HbF Absolute %HbF Change from Baseline Safety Follow-up 12 2mg FTX-6058 10 6mg FTX-6058 8 12mg FTX-6058 6 4 2 0 0 14 28 42 56 70 84 98 112 Day U.S. FDA issued a full clinical hold for FTX-6058 on February 23, 2023. Safety data collection continued with data cutoff of March 3, 2023. 4 Note: Summary data includes both subjects on and off hydroxyurea; Subject 15 ceased dosing on Day 22 and therefore, was only included in the analysis up to Day 14 Absolute %HbF Change from Baseline Mean (+/- SE)

F U L C R U M T H E R A P E U T I C S Overview of Key Inclusion and Exclusion Criteria Key Inclusion Criteria Key Exclusion Criteria Patient Severity Exclude subjects currently on Previous experience with /having received the following hydroxyurea therapies within 60 days prior to initiating FTX-6058: AND Previous experience with a stable hydroxyurea and voxelotor or dose of voxelotor or crizanlizumab or L-glutamine crizanlizumab or L-glutamine OR Lack of access to these advanced therapies We estimate that there are approximately 7,500 to 10,000 patients in the U.S. that meet the inclusion and exclusion criteria of the amended protocol 5

F U L C R U M T H E R A P E U T I C S Overview of Key Inclusion Criteria: Patient Severity Other Measures of Severity VOC ≥2 over 12 months Chronic kidney ≥2 other ≥4 over 12 disease or and at least one measures of months or ≥2 other measure of pulmonary arterial severity over 12 over 6 months hypertension severity months 6

F U L C R U M T H E R A P E U T I C S Overview of Key Inclusion Criteria: Previous Use of Hydroxyurea AND One Other Approved Therapy • Continued VOC or episodes of acute chest syndrome • Unmanageable drug-drug interactions Hydroxyurea for at least 6 months at the maximum tolerated dose • Patient refusal • Inability to tolerate the adverse effects of the therapy And • Continued pain crises and other VOCs while on stable • Inability to tolerate the adverse effects of the therapy Voxelotor or crizanlizumab dose for at least 6 months or L-glutamine • Unmanageable drug-drug interactions • Failure to increase Hb by 1 g/dL (for vox.) or continued • Patient refusal VOC episodes (for criz. or L-glutamine) Or • Lack of availability Lack of access to • Lack of insurance coverage advanced therapies 7

FTX-6058 f o r S i c k l e C e l l D i s e a s e Supplemental Detail

F U L C R U M T H E R A P E U T I C S Modifications to Key Inclusion Criteria: Patient Severity ▪ ≥4 episodes of SCD pain crisis over 12 months or ≥2 over 6 months prior to screening ▪ ≥2 episodes of SCD pain crisis plus at least one of the following over previous 12 months: • Acute chest syndrome (ACS) • Hepatic or splenic sequestration • Priapism ▪ ≥2 of the following events over the previous 12 months: • ACS • Hepatic or splenic sequestration • Priapism ▪ Tricuspid regurgitant jet velocity (TRV) ≥3.0 m/s OR TRV ≥2.5 m/s + N-terminal pro b-type natriuretic peptide (NT-proBNP) plasma level ≥160 pg/mL OR documented ongoing pulmonary hypertension diagnosed from previous echocardiogram or right-sided heart catheterization with mean pulmonary artery pressure > 25 mm Hg ▪ SCD-related chronic kidney disease (CKD) defined by at least one of the following criteria measured on 2 separate and consecutive occasions: 2 • Estimated glomerular filtration rate (eGFR) ≥30 and <60 mL/min/1.73 m • Proteinuria as defined by albumin to creatinine ratio >300 mg/g or protein to creatinine ratio >30 mg/mmol ▪ Meet medical criteria to receive (e.g., post-cerebrovascular accident) but are contraindicated for chronic transfusions (e.g., alloimmunization, transfusion reactions) 9

F U L C R U M T H E R A P E U T I C S Modifications to Key Inclusion Criteria: Previous Use of Hydroxyurea and One Other Approved Therapy Previous experience with hydroxyurea (HU) for at least 6 months at Previous experience with a stable dose of voxelotor, crizanlizumab, the maximum tolerated dose but have shown to be unresponsive or L-glutamine for at least 6 months but have shown to be and/or intolerant or ineligible based on at least one of the following unresponsive and/or intolerant or ineligible based on one of the following criteria: criteria: ▪ For voxelotor, failure to increase hemoglobin (Hb) by at least 1 ▪ Compared with baseline, same or higher number of pain crises g/dL after being on a stable dose of voxelotor for at least 6 and/or other VOCs while on HU for at least 6 months at the months maximum tolerated dose ▪ For crizanlizumab or L-glutamine compared with baseline, ▪ Episode(s) of ACS while on HU for at least 6 months at the same or higher number of pain crises and/or other VOCs while maximum tolerated dose on a stable dose of therapy for at least 6 months ▪ Inability to tolerate HU as defined by at least one of the ▪ Inability to tolerate the adverse effects of the therapy following: ▪ Unmanageable drug-drug interactions • Severe allergic reaction ▪ Lack of access to therapy • Hematologic toxicity defined by clinically significant decrease in red blood cells, white blood cells, or platelets▪ Patient refusal • Inability to tolerate gastrointestinal side effects such as nausea, vomiting, diarrhea, or abdominal pain • Inability to tolerate side effects including fatigue, dizziness, headache, fever, leg ulcers, and/or skin changes ▪ Unmanageable drug-drug interactions ▪ Patient refusal 10

F U L C R U M T H E R A P E U T I C S Summary of Exclusion Criteria ▪ Major surgery, serious illness, infection, fever, significant bleeding, cerebrovascular accident, or seizure within 14 days prior to starting study drug; elective surgery planned for the time period of the trial ▪ Sickle cell complication requiring care from a medical provider in the 14 days prior to starting study drug ▪ Use of anticoagulants or medications that induce or inhibit cytochrome P450 (CYP) 3A4, inhibit P-glycoprotein, breast cancer resistance protein, or multidrug and toxin extrusion protein 2-K, or are substrates of CYP2B6 within 14 days prior to first dose of study drug or anticipated need for any of these medications during the study. ▪ Participation in any other study with an investigational agent within the past 60 days of enrollment ▪ History of bone marrow transplant or human stem cell transplant or gene therapies ▪ Vaccination in the previous 7 days prior to initiating study drug ▪ Alanine aminotransferase ≥3× the upper limit of normal (ULN), albumin <2.0 mg/dL, direct (conjugated) bilirubin >1.5mg/dL, or prothrombin time >1.5 ULN ▪ Subjects with a history of severe renal disease defined as eGFR <30 mL/min/1.73 m2. Subjects on dialysis of any kind are excluded ▪ Subjects with abnormal laboratory results or medical history indicative of any significant medical disease that, in the opinion of the Investigator, would preclude the subject’s participation in the study or potentially obscure the interpretation of the scheduled assessments. Screening laboratory assessments may be performed twice at the discretion of the Investigator ▪ History of human immunodeficiency virus (HIV), or history of hepatitis B, or active hepatitis C. Subjects treated for and cured of hepatitis C are allowed if no longer on therapy prior to initiating study drug 11

F U L C R U M T H E R A P E U T I C S Summary of Exclusion Criteria (Continued) ▪ Subjects receiving regularly scheduled transfusions or any subject who has been transfused within 60 days prior to initiating study drug ▪ Clinically diagnosed substance use disorder for alcohol or other illicit drugs of abuse ▪ A positive urine drug screen for illicit drugs of abuse (other than opioids and marijuana/tetrahydrocannabinol [THC]/cannabidiol [CBD]) is exclusionary ▪ Pregnant or lactating female; or a female of childbearing age unable or unwilling to comply with birth control or abstinence during the study, Subject is investigative site personnel or member of their immediate family (spouse, parent, child, or sibling whether biological or legally adopted) ▪ Heart rate corrected QT interval-Fridericia’s method (QTcF) >450 msec (male) or >470 msec (female) ▪ Subject with active malignancy or history of cancer (except for squamous cell skin cancer, basal cell skin cancer, or stage 0 cervical carcinoma in situ, with no recurrence for the last 5 years) or with an immediate family member with known or suspected familial cancer syndrome. Known presence of a chromosomal abnormality or genetic mutation that may put the subject at an increased risk of myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) ▪ Subject currently on HU, voxelotor, crizanlizumab, and/ or Lglutamine or have received HU, voxelotor, crizanlizumab, and/ or L-glutamine within 60 days prior to initiating study drug 12